在单细胞RNA-seq分析中,细胞聚类分析和类型识别是核心步骤之一。通过对单细胞数据进行聚类分析,研究人员可以识别出不同的细胞群体,并对其进行细胞类型注释。随着技术的发展,聚类方法和细胞类型识别的精确度不断提高,这为生物学研究提供了更深刻的洞察。
一.细胞聚类分析与类型识别的背景:
单细胞RNA-seq技术能够捕捉到每个细胞的基因表达信息,而细胞聚类分析通过对这些数据进行模式识别,能够将具有相似表达模式的细胞归为同一类。细胞类型识别则进一步通过与已知的参考数据对比,将不同的细胞群体注释为不同的细胞类型。细胞聚类分析不仅帮助识别已知的细胞类型,还能够发现潜在的新的细胞群体和亚群体。
让生信分析更简单高效,点击图片访问【生信圆桌】获取生信云服务器

二.常用聚类算法介绍:
2.1 K-means聚类:
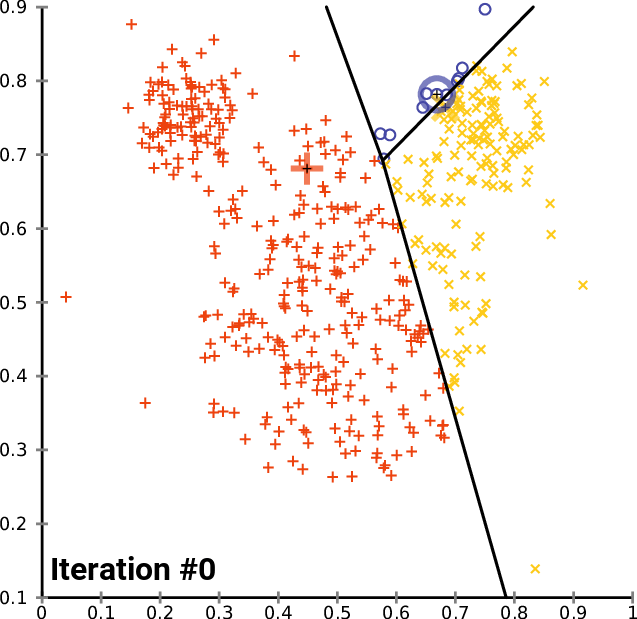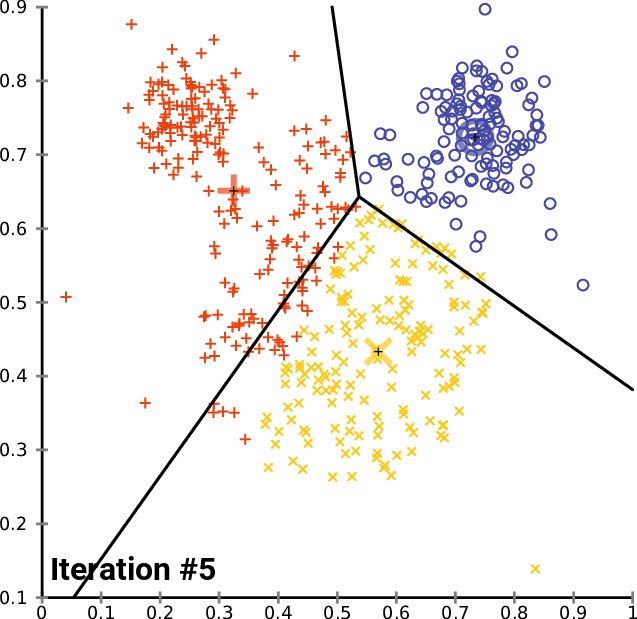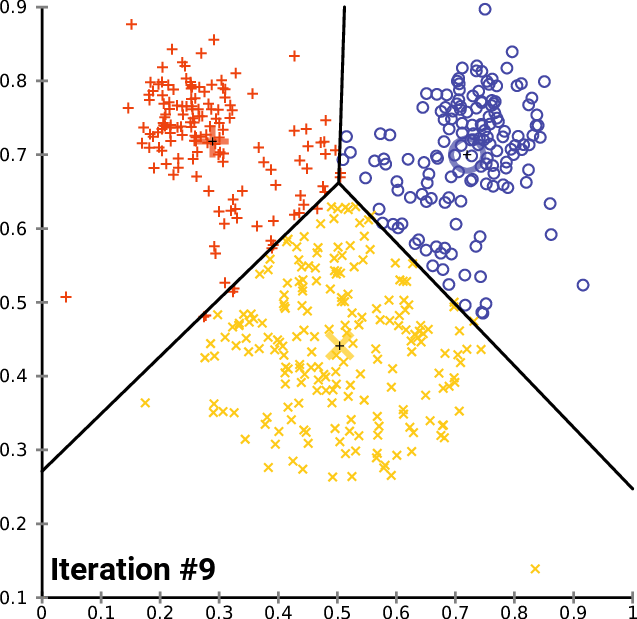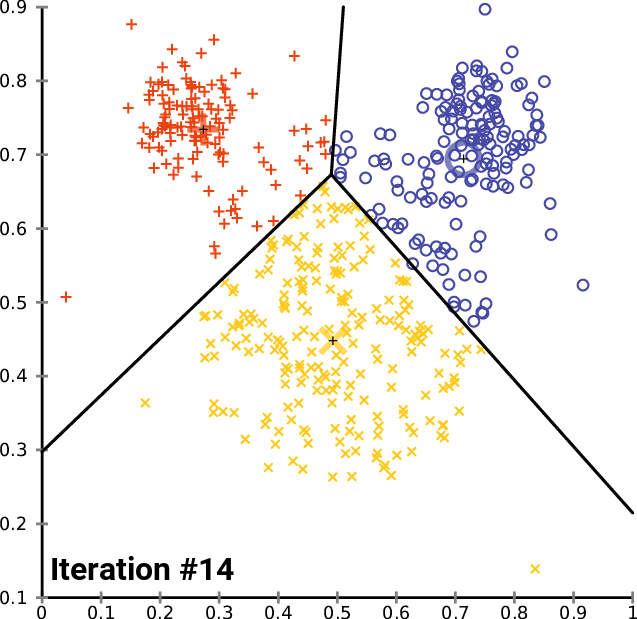
K-means聚类是最常用的聚类算法之一,通过将数据集划分为K个簇,使得每个簇内的细胞在基因表达上尽可能相似。K-means基于最小化簇内的平方误差来确定每个簇的中心点,并根据距离将数据分配到各个簇。
优点:
算法简单,计算效率高。
对大规模数据集的处理能力较强。
缺点:
需要预先指定K值,且K值的选择对于结果影响较大。
对初始聚类中心敏感,容易陷入局部最优解。
2.2层次聚类(Hierarchical Clustering):
层次聚类是基于数据间的相似性,构建出一个树状结构(树状图)。该方法不需要预先设定簇的数量,而是通过树状图的高度来决定聚类的层次,能够展示不同细胞群体间的层次关系。
优点:
不需要预先指定簇数,可以得到更多层次的聚类结果。
适用于不同大小和形状的簇。
缺点:
计算开销较大,处理大数据时效率较低。
在处理噪声数据时可能会影响聚类结果。
2.3基于密度的聚类(DBSCAN):
DBSCAN(Density-Based Spatial Clustering of Applications with Noise)是一种基于密度的聚类方法,通过在数据中寻找高密度区域来识别簇。DBSCAN能够有效处理噪声数据,并且不需要预先指定簇的数量。
优点:
不需要指定簇的数量,能够识别任意形状的簇。
对噪声和异常值具有较强的鲁棒性。
缺点:
在高维空间中效果较差,聚类结果不易解释。
对密度参数的选择敏感。
2.4自组织映射(SOM):
自组织映射(Self-Organizing Map)是一种无监督学习方法,能够将高维数据映射到二维网格中,保留数据的拓扑结构。它通过不断调整网络中的节点权重来进行聚类,适用于对细胞数据进行聚类分析。
优点:
能够在低维空间中保持高维数据的拓扑结构。
对于复杂的非线性数据也有较好的表现。
缺点:
训练过程较为复杂,需要较多的计算资源。
结果的可解释性较差。
让生信分析更简单高效,点击图片访问【生信圆桌】获取生信云服务器
三.细胞类型注释与细胞群的识别:
细胞类型注释是将聚类分析中获得的细胞群体与已知的参考数据进行对比,从而识别每个细胞群体的类型。常见的细胞类型注释方法包括:
3.1基因表达标记法:
通过已知的细胞标记基因(如T细胞标记基因CD3、B细胞标记基因CD19等),将每个聚类的细胞与已知的细胞类型进行匹配。常用的数据库有CellMarker、PanglaoDB等,它们提供了不同细胞类型的标记基因信息,帮助研究人员进行准确的注释。
3.2单细胞表达图谱(Single-cell Expression Atlas):
通过比对单细胞表达图谱中的已知细胞类型与当前数据中的聚类,进行细胞类型注释。许多公共数据库(如Human Cell Atlas)提供了详细的单细胞表达数据,可以为新的单细胞数据提供有力的参考。
3.3算法驱动的方法:
近年来,许多机器学习和深度学习算法被应用于细胞类型识别中。例如,Seurat和SingleR是常用的工具,它们能够根据表达模式和已知细胞类型进行自动注释。
四.细胞群体的识别:
细胞群体的识别不仅仅依赖于聚类算法,还需要考虑不同细胞群体之间的功能差异和生物学意义。在单细胞数据中,细胞群体的异质性往往反映了不同的生物学状态和功能。因此,聚类结果和细胞类型注释需要结合细胞的功能特征进行综合分析。
五.总结:
细胞聚类分析和类型识别在单细胞RNA-seq分析中发挥着至关重要的作用。通过聚类分析,我们能够将细胞分为具有相似表达模式的群体,而细胞类型注释则帮助我们识别这些细胞群体的生物学意义。不同的聚类算法有各自的优缺点,在实际应用中需要根据数据的特点选择最适合的算法。同时,细胞类型识别的方法不断发展,基因表达标记法、单细胞表达图谱以及机器学习方法的结合,使得细胞类型的注释更加精准。
让生信分析更简单高效,点击图片访问【生信圆桌】获取生信云服务器